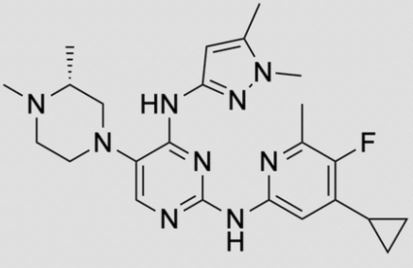
![2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=92045019&t=l)
ZY 19489, MMV 253
C24 H32 FN9, 465.5
CAS 1821293-40-6
MMV253, GTPL10024, MMV674253
N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-((3R)-2-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-3,4-dimethylpiperazin-1-yl)pyrimidin-2-amine
2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine
- N2-(4-Cyclopropyl-5-fluoro-6-methyl-2-pyridinyl)-5-[(3R)-3,4-dimethyl-1-piperazinyl]-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-2,4-pyrimidinediamine
- (R)-N2-(4-Cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine
Nature Communications (2015), 6, 6715.
https://www.nature.com/articles/ncomms7715
Hameed P., S., Solapure, S., Patil, V. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun 6, 6715 (2015). https://doi.org/10.1038/ncomms7715
Synthesis of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3, 4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (12). (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (compound 9, 190 mg, 0.42 mmol) was taken in dichloromethane (2 ml) to give a yellow suspension. To this Hunig’s Base (0.184 ml, 1.05 mmol) was added and the suspension turned clear. After 10 min of stirring, reaction mixture turned into a white suspension and then it was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 ml), and formaldehyde (0.042 ml, 0.63 mmol) was added and stirred for 10 min. To this clear solution, sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get a white suspension. The reaction mixture was concentrated and the crude product was purified through reverse-phase chromatography to get the pure off-white solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8%). Yield: 40.8%, purity: >95% by HPLC (ultraviolet at 220 and 254 nm). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s,1H), 8.03 (s, 1H) 8.00 (s, 1H) 7.67 (d, J=5.1 Hz, 1H) 6.83 (s, 1H) 3.33 (s, 3H) 2.96–2.73 (m, 4H) 2.75–2.50 (m, 1H) 2.38–2.30 (m, 4H) 2.23 (s, 7H) 2.10–1.96 (m, 1H),1.08–1.02 (m, 2H) 1.00 (d, J=6.2 Hz, 3H) 0.78–0.67 (m, 2H). 13C-NMR (126 MHz, DMO-d6) δ 155.30, 154.67, 152.10, 150.93, 148.98, 146.81. 145.29, 141.95, 140.31, 138.81, 124.91, 106.20, 97.07, 58.78, 51.87, 42.16, 35.28, 17.23. 10.99 and 8.77, HRMS (ESI): m/z calculated for C24H32FN9+H [M+H]: 466.2765. Found: 466. 2838. Traces of LC-MS, HRMS, 1H NMR and 13C-NMR of compound 12 are shown in Supplementary Figs 1–3.