 .
.Indomethacin
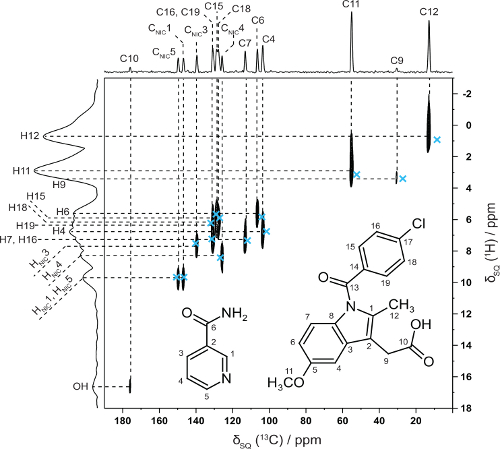
In collaboration with Professor Kenneth Harris, Cardiff University we have shown how NMR crystallography (CCPNC) can be used in synergy with structure solution by powder X-ray diffraction diffraction for an indomethacin-nicotinamide co-crystal – note the excellent 2D agreement with calculated (GIPAW, blue crosses) 13C and 1H chemical shifts for directly bonded CHs. (Dudenko 2013).

View current batch:
COA
NMR
HPLC
Datasheet
MAn improved synthesis of indole derivatives related to Indomethacin from natural Safrole
Eliezer J. Barretro*, Paulo R.R. Costa**, Perola Regina V.R. Barros and Waldemir M. Queiroz.
* Departamento de Quimica, Universidade Federal de Sao Carlos, 13.560, Sao Carlos, S.P., Brazil.
** Nucleo de Pesquisas de Produtos Naturais, Universidade Federal de Rio de Janeiro, Bl. N, 21.941, R.J., Brazil.
The indole moiety is present in a great number of natural and synthetic biologically active compounds1. Particulary in the group of non-steroidal antiinflammatory agents (NSAIA)2, among other aryl acetic acids, indomethcain 13 occupies an outstanding position. In view of the continous efforts in the preparation and pharmacological evaluation of new NSAIA4, we became interested in preparing analogues of 1, using abundant and readily accessible natural products as inexpensive starting materials. With this objective in mind, the indolyl-acetic esters 2 and 3 were chosen as initial target compounds. Using natural safrole 45 as starting material it was possible to develop under mild conditions, a synthetic sequence of high yield in which the key in the preparation of 2 and 3 is a reductive cyclization of the appropriate nitro-[beta]-keto ester 5 and 6 (Scheme 1).
In order to obtain 5 (Scheme II) the double bond of 4 was oxidized affording epoxysafrole 7 in good yields. As expected6 this compound produces the alcohol 8 (90%) by a regiospecific oxirane ring hydrogenolysis (method B). Nitration of 8 in very mild conditions 7 (HNO3)/CHCl3) gave 9 in quantitative yield. Alternatively, the alcohol 8 (93%) was synthesized by an oxymercuration-demercuration sequence8 (method A) from 4. The next step in the choosen synthetic sequence was the preparation of the methylketones 10 and 11 that was attempted by oxidation of the alcohols 8 and 9 with pyridinium chlorochromate (PCC)9 (method A) or Jones' reagent10 (method B). The nitro methylketone 12 could be C-alkylated with ethyl bromoacetate, using the intensely violet colored benzylic enolate, prepared by treatment of 12 with sodium hydride in dimethylformamide11, to furnisch the desired ester (78%). On the other hand, the lithium benzylic enolate, generated by treatment of 10 with lithium diisopropylamide (LDA) in tetrahydrofuran 12, was C-alkylated to provide the ketoesters 11 (85%) which by nitration gave 5 (98%).

Scheme I

Scheme II

Scheme III
In order to obtain the nitro-[beta]-keto esters 6, we developed the synthetic route described in Scheme III. Thus, reaction of 4 with N-bromosuccinmide (NBS) in acetone/water produces a mixture of isomeric bromohydrins 13a-c13. This mixture was treated with sodium cyanide in methanol under reflux, and the crude reaction product 14 submitted to a careful alkaline hydrolysis14 (H2O2, EtOH, 6N NaOH) gave the desired amid 15 as the sole product to crystallize as a clean colourless material from the reaction mixture. This useful sequence can be run on a 30 g scale without isolation of intermediates, in 50% overall yield. Alternatively, the bromohydrin 13a can prepared in quantitative yield by direct bromination of 7. The formation of this dibromo compound 13a can be understood by initial bromination of the 5-position of the aromatic ring followed by regioselective opening of the oxirane15 ring by the hydrobromic acid liberated in the reaction medium. A pure sample of 14 could be obtained (65%) by reaction of 13a with sodium cyanide in methanol. The conversion of 15 into hydroxy acid 16 (90%) was attempted by akaline hydrolysis. A one pot procedure for the transformation of 16 into 17 was accomplished in 75% yield by hydrogenolysis of C-Br bond over Pd/C using methanol as solvent that permits the acid catalytic esterification by action of the hydrobromic acid formed in the reaction vessel. Finally, nitration of 17 gave 18 (90%) which, when submitted to oxidation with PCC9 or Jones' reagent10 provided the nitro-[beta]-keto ester 6 in 80% yield.
With the nitro-[beta]-keto esters 5 and 6 in hand we proceeded to the last key step of the synthetic route, viz, the reductive cyclization reaction16 to provide the indole derivations 2 and 3.
So, when a methanolic solution of 5 was submitted to catalytic reduction (H2, Pd/C 60 psi) for 15 minutes at room temperature, immediately followed by solvent elimination in vacuum and subsequent "flash" chromatography filtration over silica gel, the desired indole 2 could be obtained in 80% yield, in the form of a clear oil. On the other hand, catalytic reduction of 6, under identical conditions, afforded 3 as pale yellow crystals (85%).
In conclusion, the synthetic sequence here described is useful for the preparation of indoleacetic esters starting from the allylbenzene moiety present in various abundant natural compounds such as safrole 4. The potential biologically active indole derivatives 2 and 3 can be prepared in high overall yields (46% and 20% respectively) using easily acessible reagents and mild reaction conditions.
We are indebted to the Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (40.1424/80) for financial support and fellowships to PRVRB and VMQ, to Professor Walter B. Mors (NPPN, UFRJ), for his continous encouragement and to Professor Timothy J. Srcokson (UFSCan) for his valuable suggestions.
References:
1. Pharm. Acta. Helv., 1973, 53, 65
2. Ann. Rept. Med. Chem., 1975, 10, 172, Ann Rept. Med. Chem., 1974, 9, 193
3. U.S Paten, 3, 242, 185 (Chem. Abstr., 1966, 64, 17555)
4. Inter-Alia: T.Y. Shen, J. Med. Chem., 1981, 24, 1; G.Y. Paris, D.L. Germaise, D.G. Cimon, L. Swett, G.W. Carter and P. Young, J. Med. Chem., 1980, 23, 9; D. Evans, D.W. Kunwell and T.A. Hicks, Australian (1970), 506, 085 (Chem. Abstr., 1980, 92, 169251t); M. Nagakura, T. Ota, N. Shimidzu, K. Kawamura, Y. Eto and Y. Wada, J. Med. Chem., 1979, 22, 48; J. Jeske and K. Stoskla, Acta Pol. Pharm., 1978, 35, 611.
5. W.B. Mors and C.T. Rizzini, "Botanica Economica", Epu, Sao Paulo, S.P., Brazil, 1976, p. 149.
6. R.L. Augustine, "Catalytic Hydrogenation", E. Arnold (Publishers), LTD, London and M. Dekker, Inc., New Yorik, 1965, p. 137
7. cf. D.R. Buckle; D.J. Outred, J.W. Ross., H. Smith, R.J. Smith, B.A. Spices and B.C. Gasson, J. Med. Chem., 1979, 22, 158.
8. H.C. Brown and P.G. Georghegan Jr., J. Org. Chem., 1970, 55, 1844.
9. E.J. Corey and W. Suggs, Tetrahedron Lett., 1975, 2647
10 The Jones' reagent was prepared as descripted in L.F. Fieser and M. Fieser, "Reagents for Organic Synthesis", John Wiley and Sons, Inc., New York, vol. I, p. 142
11. I.K. Stamos, Synthesis, 1980, 664
12 J.P. Albarella, J. Org. Chem., 1977, 42, 2009
13. The formation of 14c can be explained by the participation of a phenonium ion intermedite: Tetrahedron Lett., 1975, 4535.
14. C.R. Noller, "Organic Synthesis", Coll. II, John Wiley and Sons, Inc., New York, p. 586.
15. Treatment of epoxyde 7 with hydrobromic acid in acetone:water gives exclusively 4-(3-bromo-2-hydroxypropyl)-1,2-methylen
16. We have published previously the cyclization of 12 to 5,6-methylendioxy-2-methylindole in 60% yield17. Using the modified conditions described here this transformation could be accomplished in 90% yield. 17. cf. E.J. Barreiro, P.R.R. Costa, R.T. de Wello and P.R.V.R. Barros, An. Acad. brasil. Cienc., 1981, 53, 65. Experimental The 1H NMR spectra were run with Varian XL-100-12 (100 MHz) and Varian EM-360 (60 MHz) instruments, using the indicated solvent and Me4Si as internal reference. The i.r. spectra were recorded with a Perkin-Elmer 137-8 spectrophotometer, using KBr pellets. The u.v. spectra were recorded with a Beckman DGB spectrophotometer using EtOH (Uvasol) as solvent. The mass spectra were obtained with a Varian MAT-CH5-DF instrument coupled to a Varian MAT SS-100 computer system. The m.p. were determined in a Kofler apparatus. The hydrogenation reactions were performed in a Parr apparatus. Combustion analyses were carried out by CENPES - Petrobras (Rio de Janeiro, Brazil). 4-(2-Hydroxypropyl)-1,2-methylendioxy (8) Method A - Oxymercuration-Demercuration of safrole (4) Safrole (12.43 g, 76.7 mmol) was gradually added to a stirred solution of Hg(OAc)2 (25.0 g, 78.6 mmol) in THF (230 ml):H2O (75 ml). After 1 h at room temperature the yellow color was discharged and the reaction mixture was alkalinized (3M NaOH aq., 75 ml) followed by addition of a solution of NaBH4 (1.45 g, 38.15 mmol) in 3M aq. NaOH (75 ml). After 1 h the reaction mixture was saturated with NaCl, the organic layers separated and the aqeuous layer was extracted with EtOAc (4x250 ml). The combined organic layers were then washed successively with H2O (3x100 ml) and saturated brine, dried (anhy. Na2SO4) and evaporated to give 8 (12.7 g, 96%) as a clean viscous oil. An analytical sample was obtained by chromatography on silical gel using hexane-EtOAc (8:2) as eluent. Method B - Hydrogenolysis of epoxysafrole (7) A mixture of 7 (5.0 g, 27.7 mmoles) and 10% Pd/C (1.0 g) in MeOH (30 ml) was submitted to hydrogen pressure (65 psi). After 3 h the catalyst was filtered and the solvent evaporated to affort 8 (4.55 g, 90%) as a clear brown oil. Procedure for nitrations 5-Nitro-4-(2-hydroxypropyl)-1,2-methylen To a solution of 8 in CHCl3 (110 ml) was added HNO3 (d = 1.52, 3.62 ml). After 40 minutes at room temperature the reaction was neutralized with Na2CO3 aq., washed with H2O, dried (Na2SO4) and evaporated furnishing 9. (0.75 g, 100%) as yellow solid: m.p. 117-119°C from AcOH:H2O [...] 5-Nitro-4-(2-oxo-1-carboxymethylpropyl)- In a 1.5 mmol scale 11 was nitrated as described above to give as product a clear brown oil. This residue crystallized from AcOH-H2O gave 5 (80%): m.p. 96-98°C [...] 5-Nitro-4-(3-methoxycarbonyl-2-hydroxypr In a 30 mmol scale 17 was nitrated as already described to give a brown solid. Recrystallization of the crude product from AcOH:H2O gave as yellow crystals: m.p.. 106-109°C [...] PCC-Oxidations (Method A) Oxidation of 5-nitro-4-(3-methoxycarbonyl-2-hydroxypr A mixture of 18 (1.41 g, 5.0 mmol) and PCC (3.23, 15.0 mmol) in CH2Cl2 (75 ml) was allowed to react overnight at room temperature. The solvent was separated by filtration and residue washed with CH2Cl2 (2 x 50 ml). The combined organic fractions were evaporated and the residue filtered over silica gel (40 g) using CH2Cl2 as eluent, giving 6 (1.11 g, 80%) as a yellow solid: m.p. 86°C from AcOH-H2O [..] Oxidation of 4-(2-hydroxypropyl)-1,2-methylendioxyben In a 3 mmol scale the oxidation of 8 as described above followed by filtration of the crude product over silica gel gave 10 (75%) as a colorless oil: vmax 1720 cm-1, [delta]H (60 MHz, CCl4) 6.60 (3 H, narrow, m), 5.87 (2 H, s), 3.46 (2 H, s), 2.00 (3 H, s) ppm; m/z 178 (M+, 20%), 135 (100%). Oxidation of 5-nitro-4-(1-hydroxypropyl)-1,2-methylen In a 30 mmol scale the oxidation of 9 as already described gave 12 (86%) as yellow crystals: m.p. 139-141°C from AcOH-H2O [...] Jones Oxidations (Method B) Oxidation of 5-Nitro-4-(2-hydroxypropyl)-1,2-methylen To an ice cold solution of 9 (1.1 g, 4.9 mmol) in CH3COCH3 redistilled from KMnO4 (70 ml) was added dropwise a solution of Jones' reagent10 until a slight excess was present (2.0 ml), followed by addition of a saturated solution of NaHSO3 (10 ml). The organic layer was separated and the residue extracted with EtOAc (3 x 100 ml). The combined organic extrats were washed with aq. NaHCO3 (50 ml), H2O (50 ml), saturated brine, dried (Na2SO4) and evaporated. The residue crystallized from AcOH-H2O gave 12 (0.89 g, 80%). Oxidation of 4-(3-hydroxypropyl)-1,2-methylendioxyben In a 1.5 mmol scale 8 was oxidized as above giving 10 in 78% yield, after purification by preparative TLC over silica gel. Oxidation of 5-Nitro-4-(3-methoxycarbonyl-2-hydroxypr In a 10 mmol scale the Jones' oxidation of 18 was run as above affording 6 as a crystalline yellow solid crystallized from AcOH-H2O in 72% yield. |
 インドメタシン(indomethacin)の1H−NMR(D2O)スペクトルである。 【図14】
インドメタシン(indomethacin)の1H−NMR(D2O)スペクトルである。 【図14】////////
No comments:
Post a Comment