
Atom- and step-economical nucleophilic arylation of azaaromatics via electrochemical oxidative cross C-C coupling reactions
Green Chem., 2017, 19,2931-2935
DOI: 10.1039/C7GC00789B, Communication
DOI: 10.1039/C7GC00789B, Communication
O. N. Chupakhin, A. V. Shchepochkin, V. N. Charushin
A simple and efficient electrochemical method for the synthesis of asymmetrical bi(het)aryls through direct functionalization of the C(sp2)-H bond in azaaromatics with fragments of (hetero)aromatic nucleophiles has been developed
A simple and efficient electrochemical method for the synthesis of asymmetrical bi(het)aryls through direct functionalization of the C(sp2)-H bond in azaaromatics with fragments of (hetero)aromatic nucleophiles has been developed

Atom- and step-economical nucleophilic arylation of azaaromatics via electrochemical oxidative cross C–C coupling reactions
Abstract
The synthesis of asymmetrical bi(het)aryls through direct functionalization of the C(sp2)–H bond in azaaromatics with fragments of (hetero)aromatic nucleophiles has first been carried out under electrochemical oxidative conditions. This versatile method for C–C bond formation between two aryl fragments can be realized under very mild potential-controlled oxidative conditions, and it does require neither incorporation of any halogen atoms or other leaving groups, nor the use of metal catalysts. The use of the electrochemical SHN methodology for modification of azaaromatic compounds has first been demonstrated.
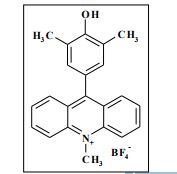




Synthesis of compounds 3a-d
The potassium tert-butoxide (0.55 mmol), corresponding phenols 2a-d (0.55 mmol) and acetonitrile (10mL) were added to electrochemical cell under argon atmosphere. The reaction mixture was stirred at room temperature for 15 min. Then 10-methylacridinium tetrafluoroborate 1 (0.5 mmol, 140 mg) was added to the reaction mixture, and stirred at room temperature for 1 h. The supporting electrolyte (40 mL) was placed in the anode and in the cathode cell compartment (10 mL). Finally, the acetic acid (1 mmol, 57 μL) was placed in the anode cell compartment. Electrolysis was carried out at a controlled potential (reference electrode Ag/AgNO3). Upon passing 2.1F of electricity (for a two-electron process), the electrolysis was stopped, the solvent was distilled off in vacuum from the anolyte, the residue was washed with 30 ml of ether and 10 ml of water. The residue was recrystallized from water and dried on air. 9-(4-Hydroxy-3,5-dimethyl-phenyl)-10-methyl-acridinium tetrafluoroborate (3a) Orange needles. 195 mg (98%). The product was identified as a compound 3a by comparing its 1H NMR spectra with its given in the literature. Satisfactory elemental analysis for C, H and N were obtained for compound 3a; none of the experimentally found percentages deviated from the theoretical values by more than 0.3%.2
1H NMR (500 MHz, [D6]DMSO): δ 9.09 (s, 1H), 8.82 (d, 2H, J=9.2 Hz), 8.45-8.42 (m, 2H), 8.12-8.10 (m, 2H), 7.94-7.91 (m, 2H), 7.16 (s, 2H), 4.90 (s, 3H), 2.33 (s, 6H) ppm. 13C NMR (126 MHz, [D6]DMSO): δ 161.5, 155.2, 141.1, 138.2, 130.3, 130.1, 127.6, 125.7, 124.8, 123.5, 119.0, 38.8, 16.6 ppm.
/////////
No comments:
Post a Comment