Concise synthesis of ketoallyl sulfones through an iron-catalyzed sequential four-component assembly
Green Chem., 2018, 20,973-977
DOI: 10.1039/C7GC03719H, Communication
DOI: 10.1039/C7GC03719H, Communication
Fuhong Xiao, Chao Liu, Dahan Wang, Huawen Huang, Guo-Jun Deng
A three starting material four component reaction (3SM-4CR) strategy is described to prepare [small beta]-acyl allylic sulfones from methyl ketones, sodium sulfinates and dimethylacetamide (DMA) in an iron-catalyzed oxidative system.
A three starting material four component reaction (3SM-4CR) strategy is described to prepare [small beta]-acyl allylic sulfones from methyl ketones, sodium sulfinates and dimethylacetamide (DMA) in an iron-catalyzed oxidative system.
Concise synthesis of ketoallyl sulfones through an iron-catalyzed sequential four-component assembly
Abstract
A three starting material four component reaction (3SM-4CR) strategy is described to prepare β-acyl allylic sulfones from methyl ketones, sodium sulfinates and dimethylacetamide (DMA) in an iron-catalyzed oxidative system. In this process, DMA was used as a dual synthon to provide two carbons. A broad range of functional groups were tolerated in this reaction system.
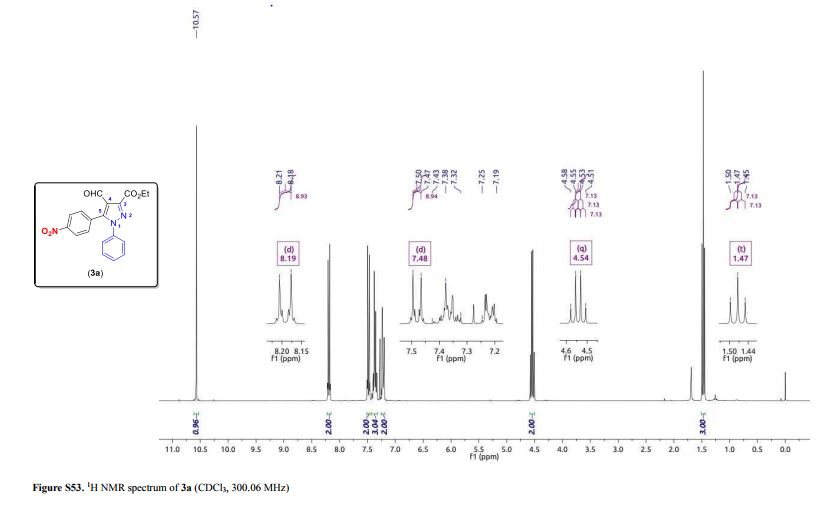
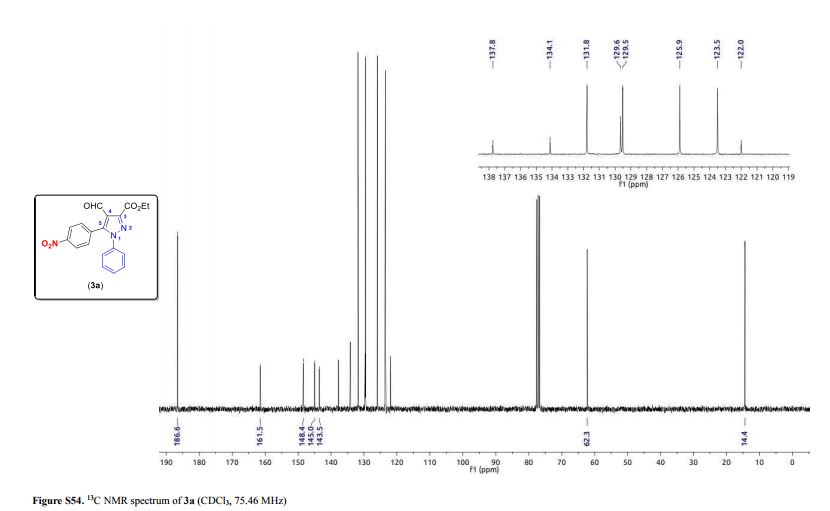
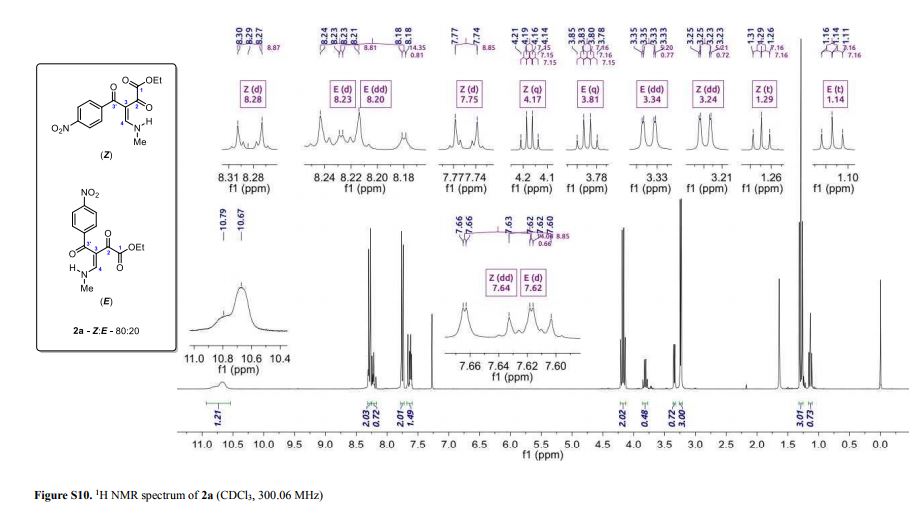
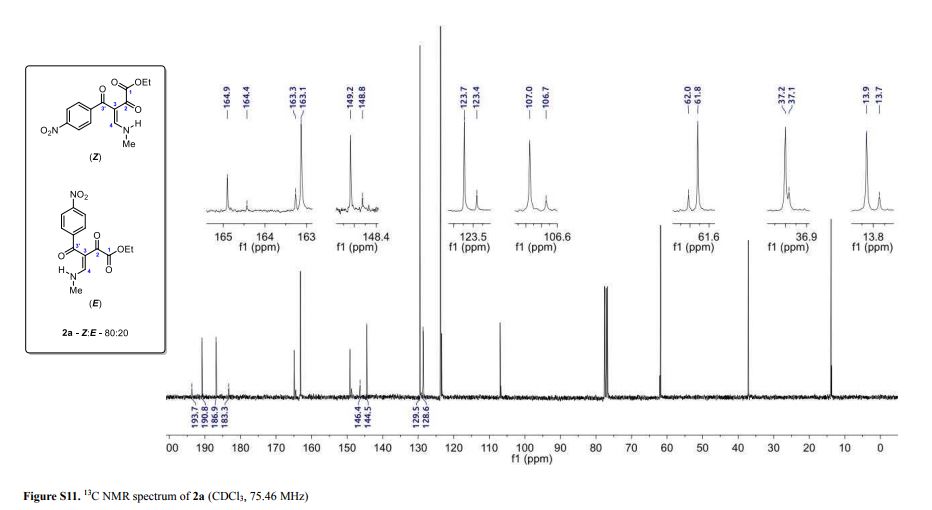
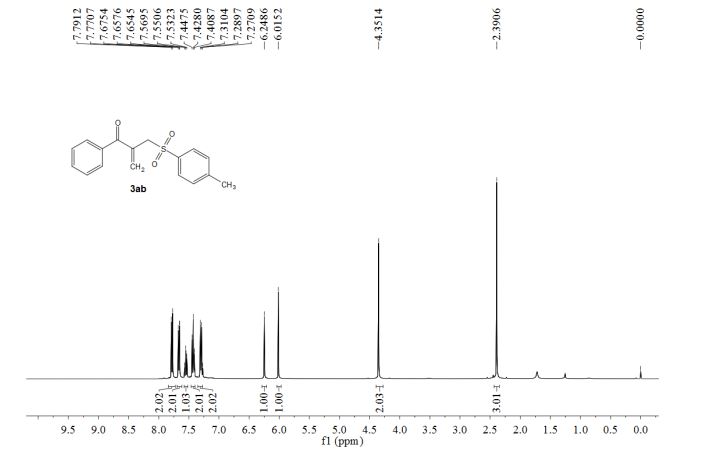
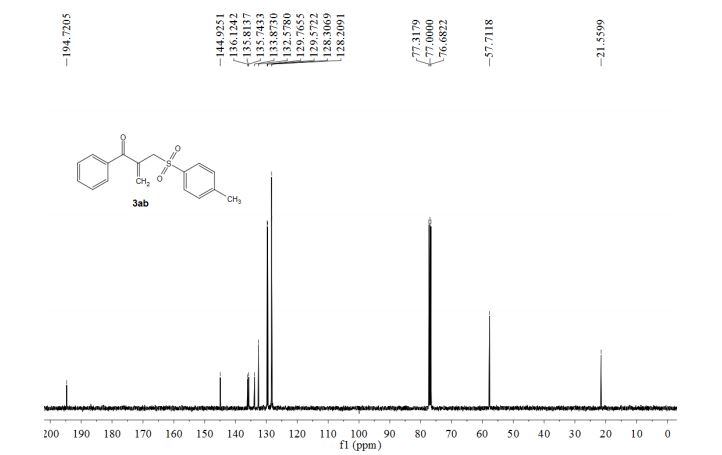
1-phenyl-2-(tosylmethyl)prop-2-en-1-one (3ab)
43.2 mg, 72% yield).
1 H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.2 Hz, 2H), 7.68-7.65 (m, 2H), 7.55 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.8 Hz, 2H), 7.30 (d, J = 8.3 Hz, 2H), 6.25 (s, 1H), 6.02 (s, 1H), 4.35 (s, 2H), 2.39 (s, 3H).
13C NMR (100 MHz, CDCl3) δ 194.7, 144.9, 136.1, 135.8, 135.7, 133.9, 132.6, 129.8, 129.6, 128.3, 128.2, 57.7, 21.6.
HRMS calcd. for: C17H17O3S+ [M+H]+ 301.08929, found 301.08908
1H NMR PREDICT




13C NMR PREDICT ABOVE
////////
Cc1ccc(cc1)S(=O)(=O)CC(=C)C(=O)c2ccccc2

![2D [1H,1H]-TOCSY, 7.4 spectrum for 4-Aminophenol](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_u3UVs02nV7755djCtgwOr6YMuxwFQ3njDRojsLwPqfhqAKyl0S2REjPuvY0cKPj9RLkGPyCboYnCi4XMrHDJiSNcvB7ho6YfuIHR7gj0FlPLbn5ij3eOwSnUOHjmdGBLKAH7v43VRoSy5jT3M7xRakKgSnqkMuYKjVSePN3LyB6lHwb-Xr5Wd-MDYAiR2RbJg8=s0-d)



![2D [1H,13C]-HSQC, 7.4 spectrum for 4-Aminophenol](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_sh1oNxvSesvBQOoiR12sdcuAIjHP_xB_rg9lpKO6Lk0by8k6QIhsRegDKZSlf_0aKNz8p6djbms6ZAzw8ln9B5VmPo-lPSA2nzZrcPfS9RI215wRACMoWFQKGk9LYsNXTABR0GnbyPEMVqWWtvq_-kEKBxqAGz3ic_zF0TQWVp-24PlG3_btC3ckymyIKC7ZMkdHk=s0-d)
![2D [1H,13C]-HMBC, 7.4 spectrum for 4-Aminophenol](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_vKWL2yjSmKi06GT76A4fRmKLu_Nrq1J_T5DehgM6FNEvkJrrm5fC_TUBZEwtWhM2G-BCXTXQ2g226uN108FwwUCiulEJweCoEL5lh9nI09vQslMWfUtGa8g4CdywLzRFpb_G_4h6mshNlwFJEkBzkGnO474fKHUbm3fsJXQsIF2m2bmPbsVG4KgZZAAiCo9ExE9UE=s0-d)