Sami E. Varjosaari, Vladislav Skrypai, Paolo Suating, Joseph J. M. Hurley, Ashley M. De Lio, Thomas M. Gilbert and Marc J. Adler
Version of Record online: 22 MAY 2017 | DOI: 10.1002/adsc.201700079
Simple Metal-Free Direct Reductive Amination Using Hydrosilatrane to Form Secondary and Tertiary Amines

Reductive amination represents the most common practical method for the synthesis of amines with the choice of a chemoselective reducing agent a key factor in determining the success of the reaction. Typically, catalytic hydrogenation, NaBH3CN or NaBH(OAc)3 are utilized, but each has specific drawbacks in terms of either toxicity or chemoselectivity. Adler et al. have reported on the use of the silane, 1-hydrosilatrane (also called silatrane), as a reducing agent for this reaction ( Adv. Synth. Catal. 2017, 359, 1872). Silatrane is easy to access, inexpensive, and air/moisture-stable and should be a good reducing agent owing to the increase in electron density at the hypervalent silicon. Model studies showed silatrane to be superior to other silanes tested with the reactions typically run neat at slightly elevated temperature. A series of scope studies showed the reaction to be both widely applicable and tolerant of many functional groups including nitro, cyano, and olefins. Extending the reaction to the formation of secondary amines necessitated the addition of AcOH to protonate the intermediate imine to facilitate reduction. The reaction was successful for both ketones and aldehydes, though failed in both cases when ammonium salts were used due to overalkylation. The reaction was demonstrated on multigram scale, and after aqueous workup, silatrane was shown to be hydrolyzed to environmentally benign products.
Synthesis of 1-hydrosilatrane (1) via boratrane [1]
To a 25 mL flask was added boric acid (50 mmol) and triethanolamine (50 mmol). Water (3 mL) was added to facilitate solubility. The flask was equipped with a short path distillation apparatus and heated to 120°C until no more water condensed. The isolated boratrane was recrystallized from acetonitrile and used directly in the next step. To an oven-dried, argon-flushed 100 mL flask containing boratrane (5 mmol) in mixed xylenes (40 mL), was added triethoxysilane (6 mmol) and anhydrous AlCl3 (0.05 mmol). The reaction was refluxed over 4 h and then cooled to room temperature. The resulting solids were filtered and further recrystallized from xylene to give silatrane as white fibrous crystals. The experimental data collected are in agreement with those described in the literature. Yield: 10.80 g (61.6 mmol, 88%); white powder; mp 257-260°C;
1H NMR (500 MHz, CDCl3) δ = 3.94 (s, 1 H), 3.83 (t, J = 6 Hz, 6 H), 2.89 (t, J = 6 Hz, 6 H);
13C NMR (125 MHz, CDCl3) 57.2, 51.2;
IR (ATR)2975, 2936, 2886, 2087, 1487, 1457, 1347, 1268, 1090, 1047, 1020, 926, 860, 748, 630, 591 cm-1 .
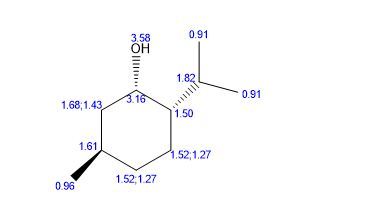
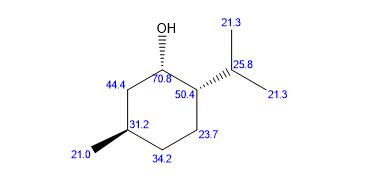
(RS)-1-Phenylethanol (3a) [3] Yield: 0.1134 g (0.9280 mmol, 93%); colourless oil.
1H NMR (500 MHz, CDCl3): δ = 7.40 (m, 4H), 7.31 (dt, J = 2.5, 7 Hz, 1H), 4.94 (q, J = 6.5 Hz, 1H), 1.54 (d, J = 6.5 Hz, 3H).
13C NMR (125 MHz, CDCl3): δ = 145.8, 128.5, 127.5, 125.4, 70.5, 25.2.
IR (ATR): 3348, 2971, 1665, 1451, 1203, 1076, 1010, 898, 759, 697, 606, 539 cm-1 .
References
[1] a) Skrypai, V.; Hurley, J. J. M.; Adler, M. J. Eur. J. Org. Chem. 2016, 2207-2211 b) Frye, C. L.; Vincent, G. A.; Finzel, W. A. J. Am. Chem. Soc. 1971, 93, 6805-6810
[2] Zelcans, G. I.; Voronkov, M. G. Chem. Heterocycl. Compd. 1967, 3, 296–298.
[3] a) Cao, L.; Ding, J.; Gao, M.; Wang, Z.; Li, J.; Wu, A. Org. Lett. 2009, 11, 3810-3813 b) Naimi-Jamal, M. R.; Mokhtari, J.; Dekamin, M. G.; Kaupp, G. Eur. J. Org. Chem. 2009, 21, 3567-3572
Main Group Metal Chemistry Vol. 23, No. 12, 2000
A NEW REACTION OF 1-HYDROSILATRANE
Edmunds Lukevics*, Luba Ignatovich, Lena Golomba, Juris Popelis, and Sergey Belyakov
Latvian Institute of Organic Synthesis , Aizkraukles 21, Riga LV1006, Latvia
< ign@osi.lv>
For the preparation of silatrane 3, a solution of oxime 1 (320 mg , 0.0017 mol) and silatrane 2 (300 mg, 0.00172 mol) in 10 ml of xylene was heated to reflux for 28 h (reaction mixture was analyzed by GC-MS every 4 h until starting products disappeared). The solvent was removed under reduced pressure, and the residue was purified by column chromatography on silicagel using a mixture of CHCI3 : CH3OH 20:1 as an eluent.